Tutorials¶
Installation¶
Introduction¶
Feature overview¶
With kmos you can:
- easily create and modify kMC models through GUI
- store and exchange kMC models through XML
- generate fast, platform independent, self-contained code [1]
- run kMC models through GUI or python bindings
kmos has been developed in the context of first-principles based modelling of surface chemical reactions but might be of help for other types of kMC models as well.
kmos’ goal is to significantly reduce the time you need to implement and run a lattice kmc simulation. However it can not help you plan the model.
kmos can be invoked directly from the command line in one of the following ways:
kmos [help] (all|benchmark|build|edit|export|help|import|rebuild|run|settings-export|shell|version|view|xml) [options]
or it may be used as an API via the kmos module.
Footnotes
| [1] | The source code is generated in Fortran90, written in a modular fashion. Python bindings are generated using f2py. |
Installation on Ubuntu Linux¶
You can fetch the current version of kmos using git
git clone http://www.github.com/mhoffman/kmos
and install it using setuptools
./setup.py install [--user]
or if you have pip run
pip install python-kmos --upgrade [--user]
To use the core functionality (programmatic model setup, code generation, model execution) kmos has a fairly modest depedency foot-print. You will need
python-numpy, a Fortran compiler, python-lxml
In order to watch the model run on screen you will additionally need
python-matplotlib, python-ase
Finally in order to use all features, in particular the GUI model editor of kmos you have to install a number of dependencies. This should not be very difficult on a recent Linux distribution with package management. So on Ubuntu it suffices to call:
sudo apt-get install gazpacho gfortran python-dev \
python-glade2 python-kiwi python-lxml \
python-matplotlib python-numpy \
python-pygoocanvas
and if you haven’t already installed it, one way to fetch the atomic simulation environment (ASE) is currently to
sudo add-apt-repository ppa:campos-dev/campos
sudo apt-get update
sudo apt-get install python-ase
Or go to their website to fetch the latest version.
Unfortunately Debian/Ubuntu have discontinued maintaining the gazpacho package which I find very unfortunate since it eased gtk GUI building a lot and I haven’t found a simple transition path (simple as in one reliable conversion script and two changed import lines) towards gtkbuilder. Therefore for the moment I can only suggest to fetch the latest old package from e.g. here and install it manually with
sudo dpkg -i gazpacho_*.deb
If you think this dependency list hurts. Yes, it does! And I am happy about any suggestions how to minimize it. However one should note these dependencies are only required for the model development. Running a model has virtually no dependencies except for a Fortran compiler.
To ease the installation further on Ubuntu one can simply run:
kmos-install-dependencies-ubuntu
Installation on openSUSE 12.1 Linux¶
On a recent openSUSE some dependencies are distributed a little different but nevertheless doable. We start by install some package from the repositories
sudo zypper install libgfortran46, python-lxml, python-matplotlib, \
python-numpy, python-numpy-devel, python-goocanvas,
python-imaging
And two more packages SUSE packages have to be fetched from the openSUSE build service
For each one just download the *.tar.bz2 files. Unpack them and inside run
python setup.py install
In the same vein you can install ASE. Download a recent version from the GitLab website unzip it and install it with
python setup.py install
Installation on openSUSE 13.1 Linux¶
In order to use the editor GUI you will want to install python-kiwi (not KIWI) and right now you can find a recent build here .
Installation on Mac OS X 10.10 or above¶
There is more than one way to get required dependencies. I have tested MacPorts and worked quite well.
- Get MacPorts
Search for MacPorts online, you’ll need to install Xcode in the process
Install Python, lxml, numpy, ipython, ASE, gcc48. I assume you are using Python 2.7. kmos has not been thoroughly tested with Python 3.X, yet, but should not be too hard.
Having MacPorts this can be as simple as
sudo port install -v py27-ipython sudo port select --set ipython py27-ipython sudo port install gcc48 sudo port select --set gcc mp-gcc48 # need to that f2py finds a compiler sudo port install py27-readline sudo port install py27-goocanvas sudo port install py27-lxml sudo port install kiwi # possibly more ... # if you install these package manually, skip pip :-) sudo port install py27-pip sudo port select --set pip pip27 pip install python-ase --user pip install python-kmos --user
Installation on windoze 7¶
In order for kmos to work in a recent windoze we need a number of programs.
Python If you have no python previously installed you should try Enthought Python Distribution (EPD) in its free version since it already comes with a number of useful libraries such a numpy, scipy, ipython and matplotlib.
Otherwise you can simply download Python from python.org and this installation has been successfully tested using python 2.7.
numpy Fetch it for your version of python from sourceforge’s Numpy site and install it. [Not needed with EPD ]
MinGW provides free Fortran and C compilers and can be obtained from the sourceforge’s MinGW site . Make sure you make a tick for the Fortran and the C compiler.
pyGTK is needed for the GUI frontend so fetch the all-in-one bundle installer and install most of it.
lxml is an awesome library to process xml files, which has unfortunately not fully found its way into the standard library. As of this writing the latest version with prebuilt binaries is lxml 2.2.8 and installation works without troubles.
ASE is needed for the representation of atoms in the frontend. So download the latest from the GitLab website and install it. This has to be installed using e.g. the powershell. So after unpacking it, fire up the powershell, cd to the directory and run
python setup.py install
in there. Note that there is currently a slight glitch in the setup.py script on windoze, so open setup.py in a text editor and find the line saying
version = ...
comment out the lines above it and hard-code the current version number.
kmos is finally what we are after, so download the latest version from github and install it in the same way as you installed ASE.
There are probably a number of small changes you have to make which are not described in this document. Please post questions and comments in the issues forum .
Installing JANAF Thermochemical Tables¶
You can conveniently use gas phase chemical potentials inserted in rate constant expressions using JANAF Thermochemical Tables. A couple of molecules are automatically supported. If you need support for more gas-phase species, drop me a line.
The tabulated values are not distributed since the terms of distribution do not permit this. Fortunately manual installation is easy. Just create a directory called janaf_data anywhere on your python path. To see the directories on your python path run
python -c"import sys; print(sys.path)"
Inside the janaf_data directory has to be a file named __init__.py, so that python recognizes it as a module
touch __init__.py
Then copy all needed data files from the NIST website in the tab-delimited text format to the janaf_data directory. To download the ASCII file, search for your molecule. In the results page click on ‘view’ under ‘JANAF Table’ and click on ‘Download table in tab-delimited text format.’ at the bottom of that page.
Todo
test installation on other platforms
Screenshots¶
The Editor¶
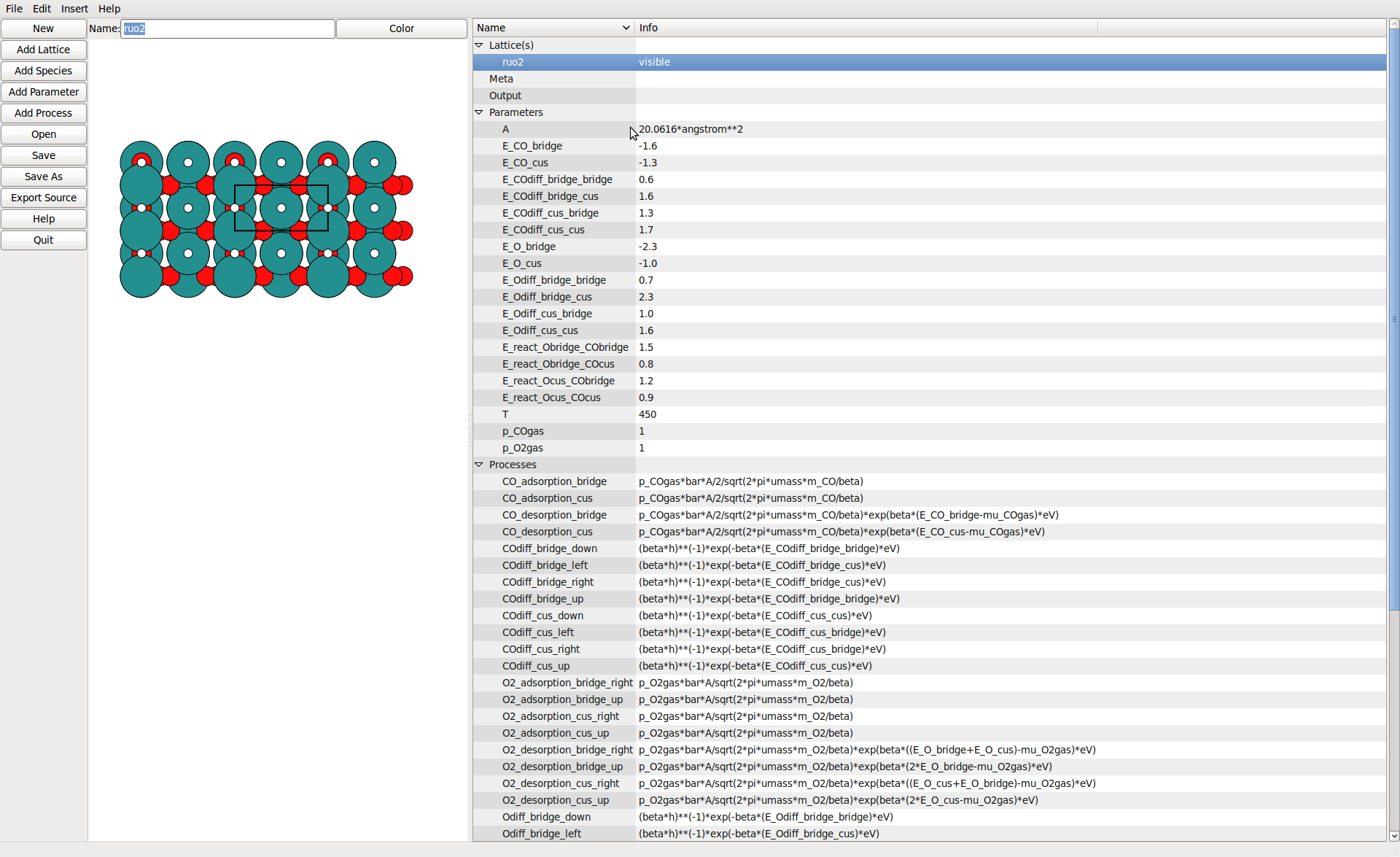
The lattice view allows to define sites by simple pointing.
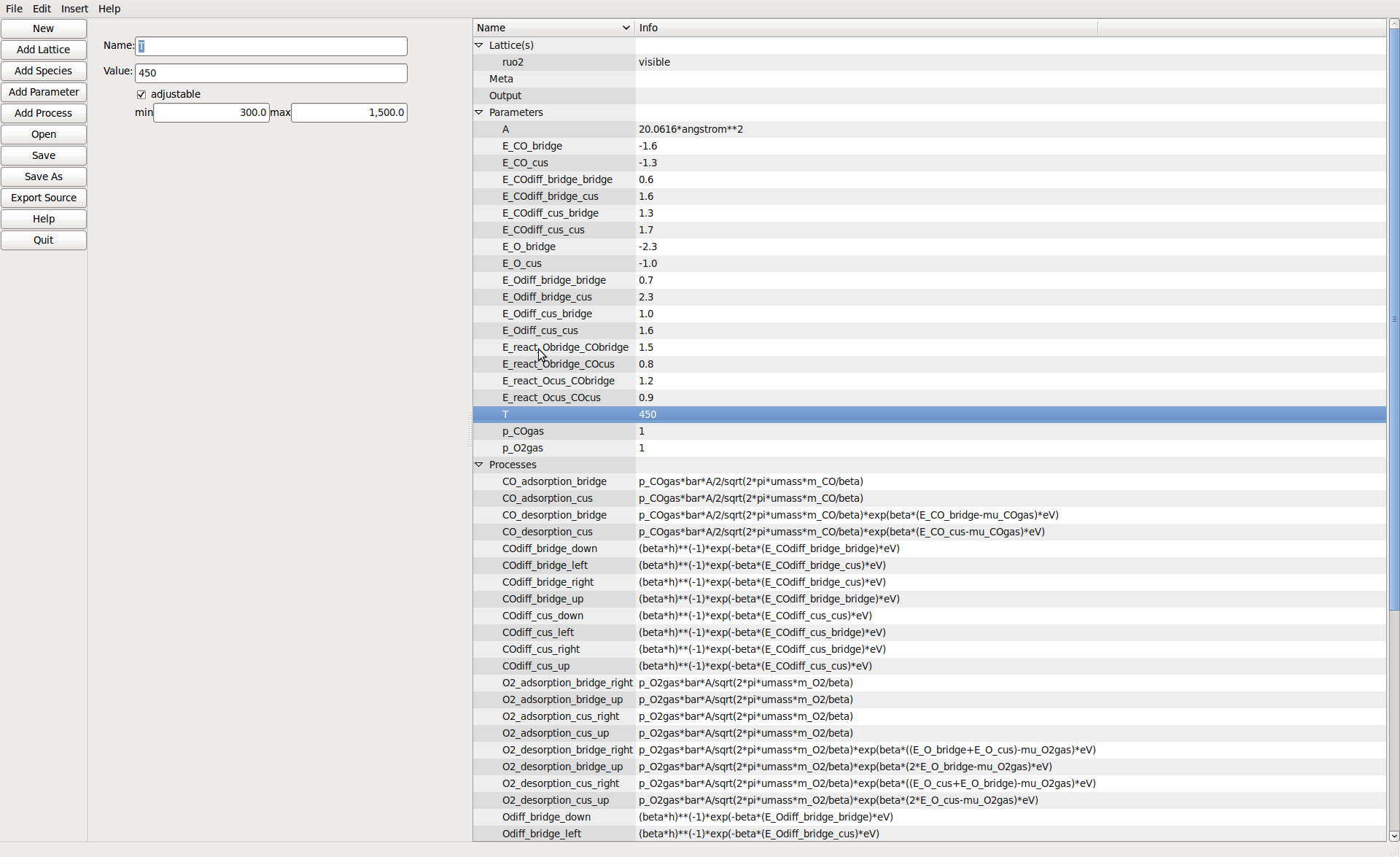
Model parameters can be defined including ranges to vary them over in the runtime viewer.
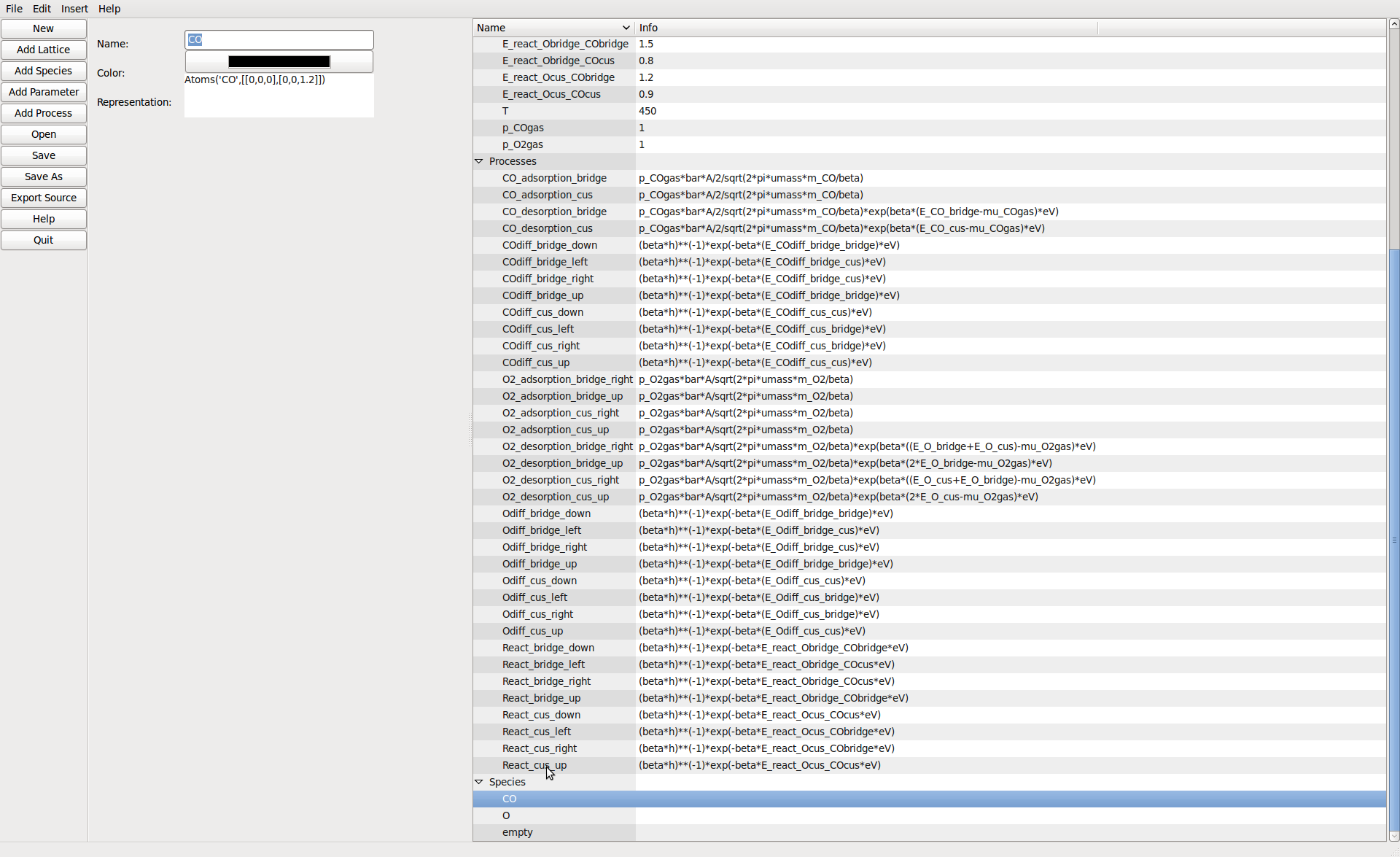
Species can be added here. The color is used to represent them in the 2D editor view. The string is an ASE atoms constructor for display at runtime.
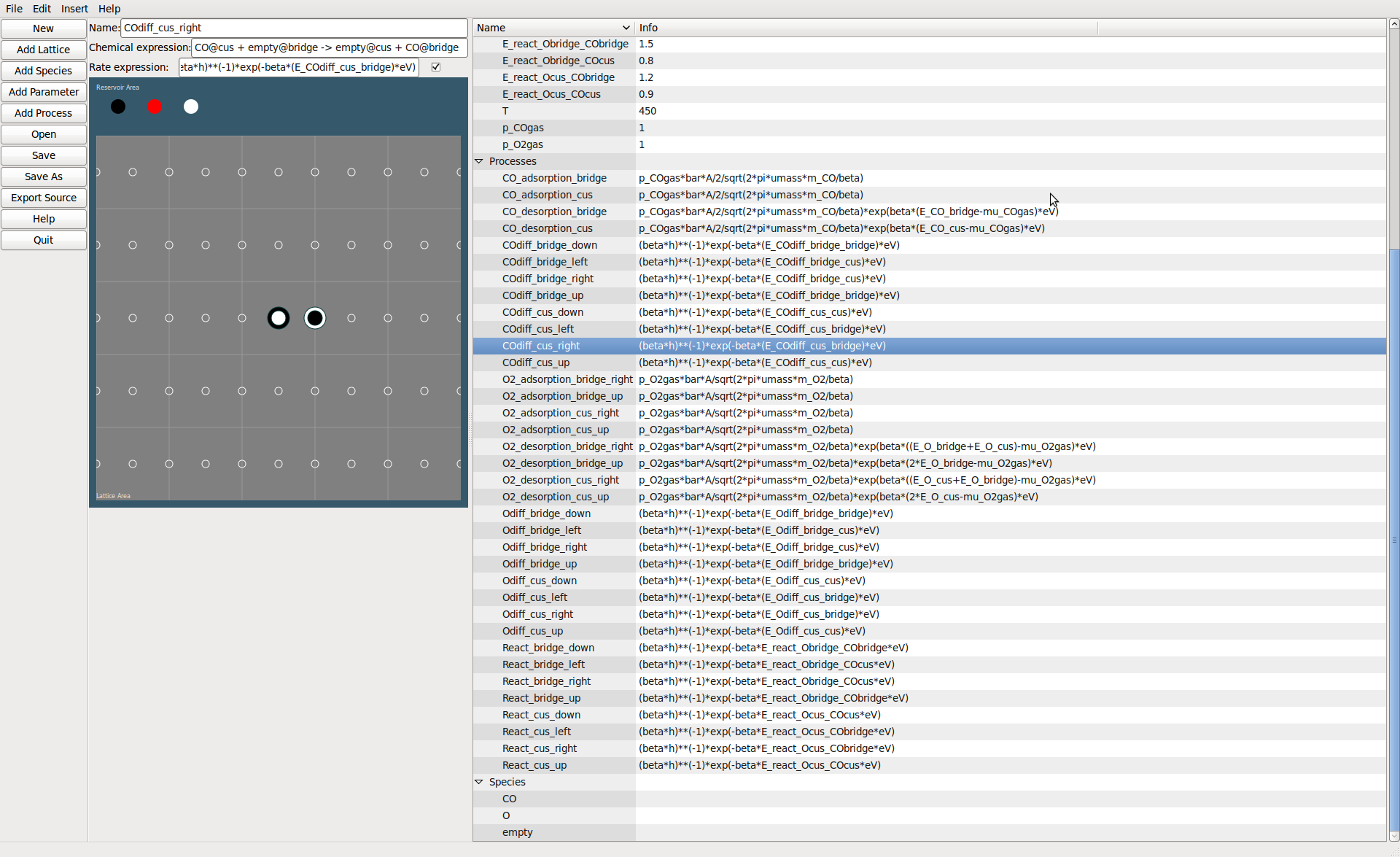
Processes can be added by point and click or by entering a chemical expression.
The Runtime View¶
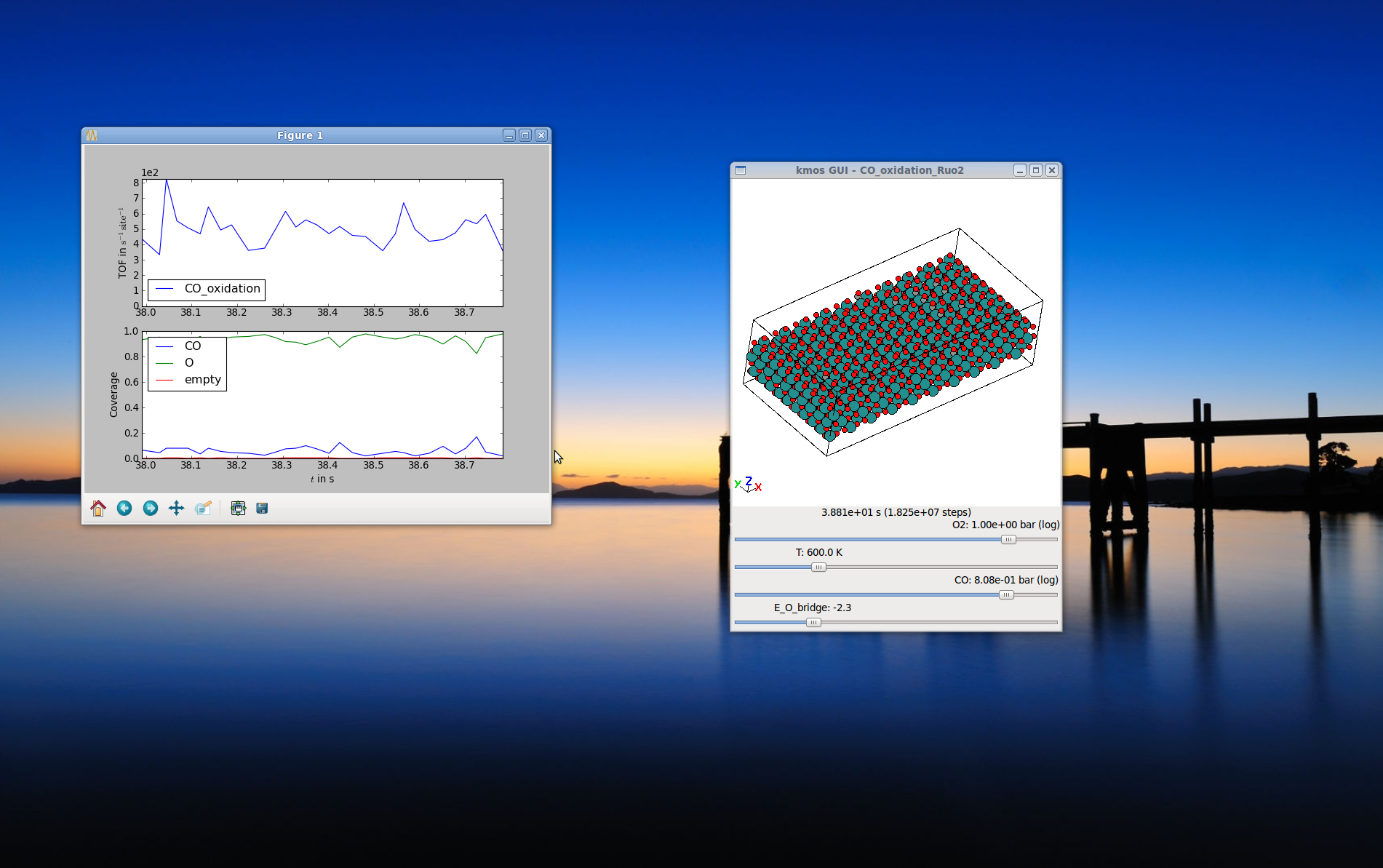
The compiled module can be run and watched in realtime. When parameters are changed this is immediately reflected in the rate constants.
A first kMC Model–the API way¶
In general there are two interfaces to defining a new model: A GUI and an API. While the GUI can be quite nice especially for beginners, it turns out that the API is better maintained simply because … well, maintaing a GUI is a lot more work.
So we will start by learning how to setup the model using the API which will turn out not to be hard at all. It is knowing howto do this will also pay-off especially if you starting tinkering with your existing models and make little changes here and there.
Construct the model¶
We start by making the necessary import statements (in *python* or better *ipython*):
from kmos.types import *
from kmos.io import *
import numpy as np
which imports all classes that make up a kMC project. The functions from kmos.io will only be needed at the end to save the project or to export compilable code.
The example sketched out here leads you to a kMC model for CO adsorption and desorption on Pd(100) including a simple lateral interaction. Granted this hardly excites surface scientists but we need to start somewhere, right?
First you should instantiate a new project and fill in meta information
pt = Project()
pt.set_meta(author = 'Your Name',
email = 'your.name@server.com',
model_name = 'MyFirstModel',
model_dimension = 2,)
Next you add some species or states. Note that whichever species you add first is the default species with which all sites in the system will be initialized. Of course this can be changed later
For surface science simulations it is useful to define an empty state, so we add
pt.add_species(name='empty')
and some surface species. Given you want to simulate CO adsorption and desorption on a single crystal surface you would say
pt.add_species(name='CO',
representation="Atoms('CO',[[0,0,0],[0,0,1.2]])")
where the string passed as representation is a string representing a CO molecule which can be evaluated in ASE namespace.
Once you have all species declared is a good time to think about the geometry. To keep it simple we will stick with a simple-cubic lattice in 2D which could for example represent the (100) surface of a fcc crystal with only one adsorption site per unit cell. You start by giving your layer a name
layer = pt.add_layer(name='simple_cubic')
and adding a site
layer.sites.append(Site(name='hollow', pos='0.5 0.5 0.5',
default_species='empty'))
Where pos is given in fractional coordinates, so this site will be in the center of the unit cell.
Simple, huh? Now you wonder where all the rest of the geometry went? For a simple reason: the geometric location of a site is meaningless from a kMC point of view. In order to solve the master equation none of the numerical coordinates of any lattice sites matter since the master equation is only defined in terms of states and transition between these. However to allow a graphical representation of the simulation one can add geometry as you have already done for the site. You set the size of the unit cell via
pt.lattice.cell = np.diag([3.5, 3.5, 10])
which are prototypical dimensions for a single-crystal surface in Angstrom.
Ok, let us see what we managed so far: you have a lattice with a site that can be either empty or occupied with CO.
Populate process list and parameter list¶
The remaining work is to populate the process list and the parameter list. The parameter list defines the parameters that can be used in the expressions of the rate constants. In principle one could do without the parameter list and simply hard code all parameters in the process list, however one looses some nifty functionality like easily changing parameters on-the-fly or even interactively.
A second benefit is that you achieve a clear separation of the kinetic model from the barrier input, which usually has a different origin.
In practice filling the parameter list and the process list is often an iterative process, however since we have a fairly short list, we can try to set all parameters at once.
First of all you want to define the external parameters to which our model is coupled. Here we use the temperature and the CO partial pressure:
pt.add_parameter(name='T', value=600., adjustable=True, min=400, max=800)
pt.add_parameter(name='p_CO', value=1., adjustable=True, min=1e-10, max=1.e2)
You can also set a default value and a minimum and maximum value set defines how the scrollbars a behave later in the runtime GUI.
To describe the adsorption rate constant you will need the area of the unit cell:
pt.add_parameter(name='A', value='(3.5*angstrom)**2')
Last but not least you need a binding energy of the particle on the surface. Since without further ado we have no value for the gas phase chemical potential, we’ll just call it deltaG and keep it adjustable
pt.add_parameter(name='deltaG', value='-0.5', adjustable=True,
min=-1.3, max=0.3)
To define processes we first need a coordinate [3]
coord = pt.lattice.generate_coord('hollow.(0,0,0).simple_cubic')
Then you need to have at least two processes. A process or elementary step in kMC means that a certain local configuration must be given so that something can happen at a certain rate constant. In the framework here this is phrased in terms of ‘conditions’ and ‘actions’. [2] So for example an adsorption requires at least one site to be empty (condition). Then this site can be occupied by CO (action) with a rate constant. Written down in code this looks as follows
pt.add_process(name='CO_adsorption',
conditions=[Condition(coord=coord, species='empty')],
actions=[Action(coord=coord, species='CO')],
rate_constant='p_CO*bar*A/sqrt(2*pi*umass*m_CO/beta)')
Note
In order to ensure correct functioning of the kmos kMC solver every action should have a corresponding condition for the same coordinate.
Now you might wonder, how come we can simply use m_CO and beta and such. Well, that is because the evaluator will to some trickery to resolve such terms. So beta will be first be translated into 1/(kboltzmann*T) and as long as you have set a parameter T before, this will go through. Same is true for m_CO, here the atomic masses are looked up and added. Note that we need conversion factors of bar and umass.
Then the desorption process is almost the same, except the reverse:
pt.add_process(name='CO_desorption',
conditions=[Condition(coord=coord, species='CO')],
actions=[Action(coord=coord, species='empty')],
rate_constant='p_CO*bar*A/sqrt(2*pi*umass*m_CO/beta)*exp(beta*deltaG*eV)')
To reduce typing, kmos also knows a shorthand notation for processes. In order to produce the same process you could also type
pt.parse_process('CO_desorption; CO@hollow->empty@hollow ; p_CO*bar*A/sqrt(2*pi*umass*m_CO/beta)*exp(beta*deltaG*eV)')
and since any non-existing on either the left or the right side of the -> symbol is replaced by a corresponding term with the default_species (in this case empty) you could as well type
pt.parse_process('CO_desorption; CO@hollow->; p_CO*bar*A/sqrt(2*pi*umass*m_CO/beta)*exp(beta*deltaG*eV)')
and to make it even shorter you can parse and add the process on one line
pt.parse_and_add_process('CO_desorption; CO@hollow->; p_CO*bar*A/sqrt(2*pi*umass*m_CO/beta)*exp(beta*deltaG*eV)')
In order to add processes on more than one site possible spanning across unit cells, there is a shorthand as well. The full-fledged syntax for each coordinate is
"<site-name>.<offset>.<lattice>"
check Manual generation for details.
Export, save, compile¶
Next, it’s a good idea to save your work
pt.filename = 'myfirst_kmc.xml'
pt.save()
Now is the time to leave the python shell. In the current directory you should see a myfirst_kmc.xml. This XML file contains the full definition of your model and you can create the source code and binaries in just one line. So on the command line in the same directory as the XML file you run
kmos export myfirst_kmc.xml
or alternatively if you are still on the ipython shell and don’t like to quit it you can use the API hook of the command line interface like
import kmos.cli
kmos.cli.main('export myfirst_kmc.xml')
Make sure this finishes gracefully without any line containining an error.
If you now cd to that folder myfirst_kmc and run:
kmos view
… and dada! Your first running kMC model right there!
If you wonder why the CO molecules are basically just dangling there in mid-air that is because you have no background setup, yet. Choose a transition metal of your choice and add it to the lattice setup for extra credit :-).
Wondering where to go from here? If the work-flow makes complete sense, you have a specific model in mind, and just need some more idioms to implement it I suggest you take a look at the examples folder. for some hints. To learn more about the kmos approach and methods you should into topic guides.
Taking it home¶
Despite its simplicity you have now seen all elements needed to implement a kMC model and hopefully gotten a first feeling for the workflow.
| [2] | You will have to describe all processes in terms of conditions and actions and you find a more complete description in the topic guide to the process description syntax. |
| [3] | The description of coordinates follows the simple syntax of the coordinate syntax and the topic guide explains how that works. |
Todo
describe modelling more complicated structures and e.g. boundary conditions
Running the Model–the GUI way¶
After successfully exporting and compiling a model you get two files: kmc_model.so and kmc_settings.py. These two files are really all you need for simulations. So a simple way to view the model is the
kmos view
command from the command line. For this two work you need to be in the same directory as these two file (more precisely these two files need to be in the python import path) and you should see an instance of your model running. This feature can be quite useful to quickly obtain an intuitive understanding of the model at hand. A lot of settings can be changed through the kmc_settings.py such as rate constant or parameters. To be even more interactive you can set a parameter to be adjustable. This can happen either in the generating XML file or directly in the kmc_settings.py. Also make sure to set sensible minimum and maximum values.
How To Prepare a Model and Run It Interactively¶
If you want to prepare a model in a certain way (parameters, size, configuration) and then run it interactively from there, there is in easy way, too. Just write a little python script. The with-statement is nice because it takes care of the correct allocation and deallocation
#!/usr/bin/env python
from kmos.run import KMC_Model
from kmos.view import main
with KMC_Model(print_rates=False, banner=False) as model:
model.settings.simulation_size = 5
with KMC_Model(print_rates=False, banner=False) as model:
model.do_steps(int(1e7))
model.double()
model.double()
# one or more changes to the model
# ...
main(model)
Or you can use the hook in the kmc_settings.py called setup_model. This function will be invoked at startup every time you call
kmos view, run, or benchmark
Though it can easily get overwritten, when exporting or rebuilding. To minimize this risk, you e.g. place the setup_model function in a separate file called setup_model.py and insert into kmc_settings.py
from setup_model import setup_model
Next time you overwrite kmc_settings.py you just need to add this line again.
Running the Model–the API way¶
In order to analyze a model in quantitatively it is more practical to write small client scripts that directly talk to the runtime API. As time passes and more of these scripts are written over and over some standard functionality will likely be integrated into the runtime API. For starters a simple script could look as follows
#!/usr/bin/env python
from kmos.run import KMC_Model
model = KMC_Model()
An alternative way that gets you started fast it to run
kmos shell
and just interact directly with model. It is often a good idea to
%logstart some_scriptname.py
first in the IPython command to save what you have typed for later use.
As you can see by default the model prints a disclaimer and all rate constants, which can each be turned off by instantiating
model = KMC_Model(print_rates=False, banner=False)
The most important method is of course how to run the model, which you can do by saying
model.do_steps(100000)
which would run the model by 100,000 kMC steps.
Let’s say you want to change the temperature and a partial pressure of the model you could type
model.parameters.T = 550
model.parameters.p_COgas = 0.5
and all rate constants are instantly updated. In order get a quick overview of the current settings you can issue e.g.
print(model.parameters)
print(model.rate_constants)
or just
print(model)
Now an instantiated und configured model has mainly two functions: run kMC steps and report its current configuration.
To analyze the current state you may use
atoms = model.get_atoms()
Note
If you want to fetch data from the current state without actually visualizing the geometry can speed up the get_atoms() call using
atoms = model.get_atoms(geometry=False)
This will return an ASE atoms object of the current system, but it also contains some additional data piggy-backed such as
model.get_occupation_header()
atoms.occupation
model.get_tof_header()
atoms.tof_data
atoms.kmc_time
atoms.kmc_step
These quantities are often sufficient when running and simulating a catalyst surface, but of course the model could be expanded to more observables. The Fortran modules base, lattice, and proclist are atttributes of the model instance so, please feel free to explore the model instance e.g. using ipython and
model.base.<TAB>
model.lattice.<TAB>
model.proclist.<TAB>
etc..
The occupation is a 2-dimensional array which contains the occupation for each surface site divided by the number of unit cell. The first slot denotes the species and the second slot denotes the surface site, i.e.
occupation = model.get_atoms().occupation
occupation[species, site-1]
So given there is a hydrogen species in the model, the occupation of hydrogen across all site type can be accessed like
hydrogen_occupation = occupation[model.proclist.hydrogen]
To access the coverage of one surface site, we have to remember to subtract 1, when using the the builtin constants, like so
hollow_occupation = occupation[:, model.lattice.hollow-1]
Lastly it is important to call
model.deallocate()
once the simulation if finished as this frees the memory allocated by the Fortan modules. This is particularly necessary if you want to run more than one simulation in one script.
Generate Grids of Sampled Data¶
For some kMC applications you simply require a large number of data points across a set of external parameters (phase diagrams, microkinetic models). For this case there is a convenient class ModelRunner to work with
from kmos.run import ModelRunner, PressureParameter, TemperatureParameter
class ScanKinetics(ModelRunner):
p_O2gas = PressureParameter(1)
T = TemperatureParameter(600)
p_COgas = PressureParameter(min=1, max=10, steps=40)
ScanKinetics().run(init_steps=1e8, sample_steps=1e8, cores=4)
This script generates data points over the specified range(s). The temperature parameters is uniform grids over 1/T and the pressure parameters is uniform over log(p). The script can be run synchronously over many cores as long as the cores can access the same file system. You have to test whether the steps before sampling (init_steps) as well as the batch size (sample_steps) is sufficient.
Manipulating the Model at Runtime¶
It is quite easy to change not only model parameters but also the configuration at runtime. For instance if one would like to prepare a surface with a certain configuration or pattern.
Given you instantiated a model instance a site occupation can be changed by calling
model.put(site=[x,y,z,n], model.proclist.<species>)
However if changing many sites at once this is quite inefficient, since each put call, adjusts the book-keeping database. To circumvent this you can use the _put method, like so
model._put(...)
model._put(...)
...
model._adjust_database()
though at the end one must not forget to call _adjust_database() before executing any next step or the database of available processes is inaccurate and the model instance will crash soon.
You can also get or set the whole configuration of the lattice at once using
config = model._get_configuration()
# possible change config
model._set_configuration(config)
Running models in parallel¶
Due to the global clock in kMC there seems to be no simple and efficient way to parallelize a kMC program. kmos certainly cannot parallelize a single system over processors. However one can run several kmos instances in parallel which might accelerate sampling or efficiently check for steady state conditions.
However in many applications it is still useful to run several models seperately at once, for example to scan some set of parameters one a multicore computer. This kind of problem can be considered embarrasingly parallel since it requires no communication between the runs.
This is made very simple through the multiprocessing module, which is in the Python standard library since version 2.6. For older versions this needs to be downloaded <http://pypi.python.org/pypi/multiprocessing/> and installed manually. The latter is pretty straightforward.
Then besides kmos we need to import multiprocessing
from multiprocessing import Process
from numpy import linspace
from kmos.run import KMC_Model
and let’s say you wanted to scan a range of temperature, while keeping all other parameteres constant. You first define a function, that takes a set of temperatures and runs the simulation for each
def run_temperatures(temperatures):
for T in temperatures:
model = KMC_Model()
model.parameters.T = T
model.do_steps(100000)
# do some evaluation
model.deallocate()
In order to split our full range of input parameters, we can use a utility function
from kmos.utils import split_sequence
All that is left to do, is to define the input parameters, split the list and start subprocesses for each sublist
if __name__ == '__main__':
temperatures = linspace(300, 600, 50)
nproc = 8
for temperatures in split_sequence(temperatures, nproc):
p = Process(target=run_temperatures, args=(temperatures, ))
p.start()
Development¶
Contributions of any sort are of course quite welcome. Patches and comments are ideally sent in form of email, pull request, or github issues.
To make synergizing a most pleasing experience I suggest you use git, nose, pep8, and pylint
sudo apt-get install git python-nose pep8 pylint
When sending a patch please make sure the nose tests pass, i.e. run from the top project directory
nosetests
To make testing and comparison even easier it would be helpful if you create an account with Travis CI and run your commits through the test suite.
Have a look at Google’s Python style guide as far as style questions go.